What is Usher syndrome?
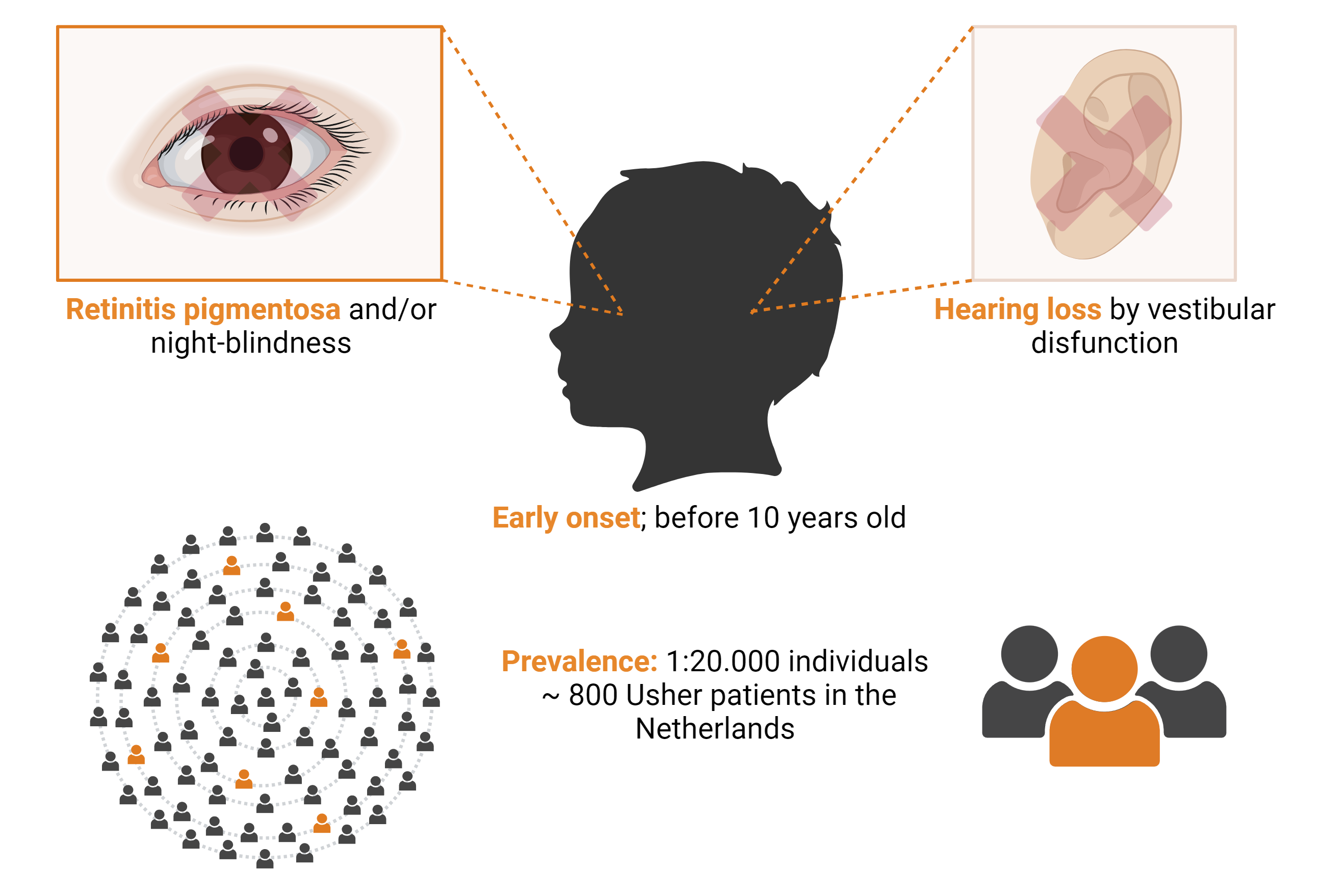
Usher syndrome is the most common cause of hereditary deaf-blindness in man. It is characterized by congenital hearing loss, a progressive loss of vision as a consequence of retinitis pigmentosa, occasionally also accompanied by vestibular dysfunction. The initial symptom of retinitis pigmentosa is often an impaired ability to see in dim-light situations (night-blindness). Retinitis pigmentosa is further characterized by a progressive loss of peripheral vision, resulting in tunnel vision, and eventually also affecting central vision. The general prevalence of Usher syndrome is 1:20.000 individuals, resulting in approximately 800 Usher syndrome patients in the Netherlands.
What causes Usher syndrome?
Pathogenic variants in so far 11 different genes have been identified as the underlying cause of Usher syndrome. Mutations in the USH2A gene are most prevalent, explaining approximately 50% of all Usher syndrome cases. Besides resulting in Usher syndrome type 2, mutations in USH2A are also the primary cause of recessively inherited non-syndromic retinitis pigmentosa. USH2A encodes the large transmembrane protein usherin, that together with all other USH1 and USH2 proteins functions in a highly dynamic protein network in both retina and inner ear. This protein network is important for providing structural support to retinal photoreceptors and inner ear hair cells, and it has been proposed to be involved in regulating the transport of photopigments from the photoreceptor inner segments towards the outer segments. Absence of usherin thus results in the aberrant localization of photopigments, leading to the highly toxic ectopic phototransduction. As a consequence, photoreceptors will degenerate resulting in vision loss.
What types of Usher syndrome can be distinguished?
Usher syndrome can be clinically divided into four distinct types based on the severity and progression of the hearing loss, the age of onset of the visual dysfunction and the presence or absence of vestibular dysfunction:
- Usher syndrome type 1 (USH1) is characterized by profound congenital hearing loss, vestibular dysfunction and onset of retinitis pigmentosa before puberty. So far, pathogenic variants in six different genes have been identified to underly USH1 (MYO7A, USH1C, CDH23, PCDH15, USH1G and CIB2).
- Usher syndrome type 2 (USH2) is characterized by congenital hearing impairment, normal vestibular function and onset of retinitis pigmentosa shorty after puberty. Variants in USH2A, ADGRV1 and WHRN have been identified to be associated with USH2.
- Usher syndrome type 3 (USH3) is characterized by progressive hearing loss, a variable onset of retinitis pigmentosa, occasionally with vestibular dysfunction. USH3 is caused by pathogenic variants in the CLRN1
- Usher syndrome type 4 (USH4) is characterized by late-onset progressive hearing loss and late-onset retinitis pigmentosa. Pathogenic variants in the ARSG gene are the underlying cause of USH4.